library(diseasy)
#> Loading required package: diseasystore
#>
#> Attaching package: 'diseasy'
#> The following object is masked from 'package:diseasystore':
#>
#> diseasyoptionOverview
The DiseasyRegions module defines the regional scope for
the models. In addition, it provides a common place to define the
demography available for those regions, and the adjacency between
regions.
The module is intentionally generic. Region identifiers are treated
as ordinary character values and are matched exactly. This means that,
e.g., the region "north" is not interpreted as a parent of
"north_subregion".
Data structure
DiseasyRegions uses three inputs:
-
regions: acharactervector defining the selected region scope. -
demography: adata.framewith one region column, optional stratification columns, and one population column. -
adjacency: adata.framedescribing connectedness between pairs of regions (see the Regional adjacency section for further details).
The demography data must include the columns region and
population. Any additional columns are treated as
stratification variables.
demography_nordic
#> # A tibble: 505 × 3
#> region age population
#> <chr> <dbl> <dbl>
#> 1 DK 0 60965
#> 2 DK 1 61357
#> 3 DK 2 61663
#> 4 DK 3 62128
#> 5 DK 4 61148
#> # ℹ 500 more rowsThe adjacency data must include exactly the columns
from, to, and adjacency. The
from and to columns should contain the same
set of region identifiers.
adjacency_meta_nordic
#> # A tibble: 25 × 3
#> from to adjacency
#> <chr> <chr> <dbl>
#> 1 NO NO 6413.
#> 2 NO FI 13.7
#> 3 FI NO 13.7
#> 4 SE NO 52.1
#> 5 NO SE 52.1
#> # ℹ 20 more rowsRegional adjacency
DiseasyRegions builds on the concept of a degree of
“connectedness” between the different regions. Here, this
“connectedness” is referred to as “adjacency” in alignment with other
models.
Different measures for adjacency can be extracted from real-world data which have very different interpretations. Adjacency is often described from information about “movement” such as mobility data or surveys on how people move between regions. Alternatively adjacency can be derived from “infection” data such as genetic phylogeny.
While these seem very similar, there are substantial differences in
how these adjacencies are expressed numerically, and how they should be
implemented in disease spread models. Our framework assumes a reality
where disease is transported between regions by people returning to
their own region (travel or commuting), that is: there is no immigration
or emigration. If the supplied adjacency describes a
movement matrix it is furthermore assumed that people travelling
to a place transmits disease equally with people travelling
from a place. Which in turns means that the resulting infection
matrix used in the models will be symmetric.
In diseasy, the adjacency of DiseasyRegions
are implemented in SEIR-like models and it helps to be clear on how this
adjacency is used to illustrate these differences.
To begin, let’s define the “movement matrix” between connected regions:
If we interpret these adjacencies as “the average fraction of contacts between regions”, we can diagram the SEIR compartments of a two-region model as:
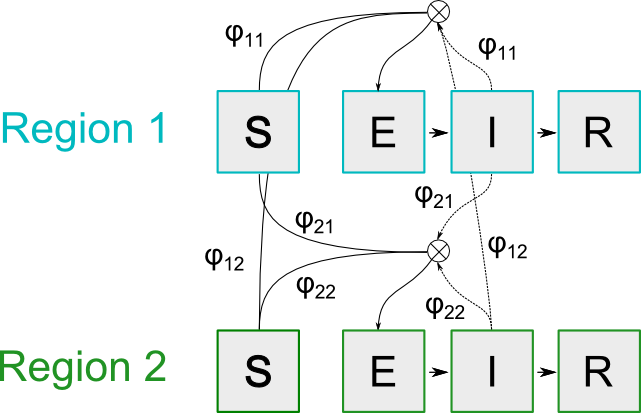
SEIR model overview for a two region model
With the following equations for the infectious compartments in region where we divide the types of contacts into whether individuals stay in their own region or travel1 to other regions.
In general, we can express the dynamics in matrix form assuming different regions
Where is the Hadamard product (element-wise multiplication).
The matrix elements of contains the probability that individual interact across regions, with elements .
These elements compute the probability that individuals in region travel to region AND individuals in region travel to region .
For the 2-region system, the mixing can be expressed in matrix form as:
In this formalism, the which satisfies:
requires
That leads to the following expression: $$ S_1 (\phi_{1,1}^2 + \phi_{2,1}^2) I_1 + S_1 (\phi_{1,1}\phi_{1,2} + \phi_{2,1}\phi_{2,2}) I_2 + \\ S_2 (\phi_{2,2}^2 + \phi_{1,2}^2) I_2 + S_2 (\phi_{2,2}\phi_{2,1} + \phi_{1,2}\phi_{1,1}) I_1 $$
If we assume there is no spatial structure and therefore all matrix elements are identical:
Then we can reduce the expression
Which means that the neutral matrix elements for the 2-region system are:
and in general for the n-region system2:
This “movement matrix” can be estimated from information on how individuals travel, such as from survey or mobility data.
Alternatively, we can consider the matrix which we can think of as a “infection-flow” matrix. This matrix identifies the flow of infections from individuals residing in regions to individuals residing in regions . Such matrix may be estimated from more direct measurements of disease flow such as from genetic data or contact tracing.
The matrix will be normalised such that its row sums are 1.
DiseasyRegions can take either type of adjacency
matrix.
The stored adjacency input is long form, but the module also exposes
an $infection_flow_matrix field. This field
returns a matrix representation of adjacency for the currently selected
regions.
The matrix is derived in three steps:
- the stored adjacency data are filtered to the selected regions;
- the matrix is normalised so that row sums are 1.
Creating a regional module
A DiseasyRegions object can be created and then
configured with the selected regions, demography, and adjacency data.
Each setter validates the candidate input before storing it.
region <- DiseasyRegions$new()
region
#> # DiseasyRegions #############################################
#> Regions: No regions have been specified
#> Total population: No population data loaded
#> Theta matrix: No adjacency data loaded
region$set_regions(regions = c("DK", "SE", "NO", "FI"))
region$set_demography(demography = demography_nordic)
region$set_adjacency(adjacency = adjacency_meta_nordic)
region
#> # DiseasyRegions #############################################
#> Regions: DK, FI, NO, SE
#> Total population: 27,124,322
#> Theta matrix: Max eigenvalue 1.01The $demography field returns demography
filtered to the currently selected regions.
region %.% demography
#> # A tibble: 404 × 3
#> region age population
#> <chr> <dbl> <dbl>
#> 1 DK 0 60965
#> 2 DK 1 61357
#> 3 DK 2 61663
#> 4 DK 3 62128
#> 5 DK 4 61148
#> # ℹ 399 more rowsPopulation totals can be computed directly from the filtered demography.
aggregate(
population ~ region,
data = region %.% demography,
FUN = sum
)
#> region population
#> 1 DK 5837230
#> 2 FI 5522887
#> 3 NO 5381527
#> 4 SE 10382678The active bindings $adjacency and
$infection_flow_matrix expose derived views
of adjacency input after filtering to the selected
regions.
region %.% adjacency
#> # A tibble: 16 × 3
#> from to adjacency
#> <chr> <chr> <dbl>
#> 1 DK DK 9753.
#> 2 DK FI 11.5
#> 3 DK NO 75.9
#> 4 DK SE 39.1
#> 5 FI DK 11.5
#> 6 FI FI 3512.
#> 7 FI NO 13.7
#> 8 FI SE 24.2
#> 9 NO DK 75.9
#> 10 NO FI 13.7
#> 11 NO NO 6413.
#> 12 NO SE 52.1
#> 13 SE DK 39.1
#> 14 SE FI 24.2
#> 15 SE NO 52.1
#> 16 SE SE 1360.
region %.% infection_flow_matrix
#> DK FI NO SE
#> DK 0.974618393 0.004387903 0.018988342 0.03012657
#> FI 0.004387903 0.972520508 0.005932881 0.02263160
#> NO 0.018988342 0.005932881 0.957411341 0.04222013
#> SE 0.030126573 0.022631597 0.042220133 0.85190061The module configuration can also be visualised via the
$plot() method with or plot().
plot(region)
#> Linking to GEOS 3.12.1, GDAL 3.8.4, PROJ 9.4.0; sf_use_s2() is TRUERegional module with hierarchical structure (NUTS)
If you have demography and adjacency data for structured regions
(such as NUTS), then DiseasyRegionsNuts can handle the
nested data.
region_nuts <- DiseasyRegionsNuts$new()
region_nuts
#> # DiseasyRegions #############################################
#> Regions: No regions have been specified
#> Total population: No population data loaded
#> Theta matrix: No adjacency data loaded
region_nuts$set_regions(regions = "DK")
region_nuts$set_demography(demography = demography_nordic_nuts3)
region_nuts$set_adjacency(adjacency = adjacency_meta_nordic_nuts)
region_nuts
#> # DiseasyRegions #############################################
#> Regions: DK
#> Total population: 5,992,734
#> Theta matrix: Max eigenvalue 1.07And again the module configuration can also be visualised with
plot().
plot(region_nuts)Changing the selected regions
The selected regional scope can be changed with
$set_regions(). The new regions must be available in the
stored adjacency and demography data.
The active fields now reflect the updated scope.
region %.% demography
#> # A tibble: 202 × 3
#> region age population
#> <chr> <dbl> <dbl>
#> 1 DK 0 60965
#> 2 DK 1 61357
#> 3 DK 2 61663
#> 4 DK 3 62128
#> 5 DK 4 61148
#> # ℹ 197 more rows
region %.% adjacency
#> # A tibble: 4 × 3
#> from to adjacency
#> <chr> <chr> <dbl>
#> 1 DK DK 9753.
#> 2 DK SE 39.1
#> 3 SE DK 39.1
#> 4 SE SE 1360.
region %.% infection_flow_matrix
#> DK SE
#> DK 0.99203703 0.03174343
#> SE 0.03174343 0.94562542Validation
The module checks that selected regions are available in the stored demography and adjacency. For example, trying to select a region that is not present in the data will fail.
try(region$set_regions(regions = "DE"))
#> Error in base::tryCatch(base::withCallingHandlers({ :
#> `regions` and `adjacency` must contain at least one common region.The adjacency data must also contain the same region identifiers in
the from and to columns.
bad_adjacency <- data.frame(
from = c("DE", "BE"),
to = c("DE", "BE"),
adjacency = c(1, 1)
)
try(region$set_adjacency(adjacency = bad_adjacency))
#> Error in base::tryCatch(base::withCallingHandlers({ :
#> `regions` and `adjacency` must contain at least one common region.